
Current Version: Profex 5.5.0 - Released May 17, 2025

Disclaimer: This tutorial provides technical information on how to combine calibrated peak lists with a Rietveld refinement project in Profex/BGMN. The method was introduced by Taylor and Rui [1] and Taylor and Matulis [2], and is sometimes referred to as PONKCS [3]. The accuracy may strongly vary from sample to sample and depend on the complexity of the phase in question, and whether or not it is available in pure form. Assessing the suitability of the method and the quality of the results remains up to the user.
Example files
The example datasets contain only raw data. Use these files to reproduce the step-by-step instructions given below.
Complete refinement projects contain all the results obtained by following the instructions below.
| Files | Windows | Mac/Linux |
| Example datasets (90 kb) | Tutorial-Fhkl-examples-win-200419.zip | Tutorial-Fhkl-examples-unix-200419.zip |
| Complete refinement projects / solutions (600 kb) | Tutorial-Fhkl-solutions-win-200419.zip | Tutorial-Fhkl-solutions-unix-200419.zip |
Introduction
Rietveld refinement requires complete structure descriptions for all crystalline phases in order to determine relative phase quantities. If a phase is present of which only the space group and unit cell dimensions but no atomic coordinates are known, it can be added to the refinement and processed in LeBail mode. This improves the fit because no unassigned peaks interfere with the fit of the assigned peaks, but the LeBail-fitted phase cannot be quantified due to missing essential information on the composition and structure.
In a very basic Rietveld refinement, the algorithm reads the structural information of all phases and calculates peak positions from the cell parameters and the space group. Then it calculates structure factors from the atomic sites, converts them to intensities and convolutes various profile parameters. The as-obtained peaks are then fitted to the observed peaks: The position on the 2θ axis is matched by optimizing the cell parameters, and the intensity is matched by changing the scale factor S, which scales all peak intensities up or down. Once the fit converges, the relative weight fraction of phase a is then calculated from the optimized scale factors S, the optimized cell parameters (i.e. the unit cell volume V), and other structural parameters as follows:
![\[ W_a = \frac{S_a \cdot (Z \cdot M \cdot V)_a}{\sum S_i \cdot (Z \cdot M \cdot V)_i} \]](https://www.profex-xrd.org/wp-content/ql-cache/quicklatex.com-756dee344ef40a1025fe53ec32bcc4e0_l3.png "Rendered by QuickLaTeX.com")
W is the relative weight fraction, S is the Rietveld scale factor, Z is the number of formula units per unit cell, M is the mass of the formula unit, and V is the volume of the unit cell.
If the atomic site information is unknown, it is possible to take a shortcut. Instead of letting the Rietveld algorithm calculate structure factors from the atomic site information, we can provide a list of pre-calculated structure factors. The algorithm then continues from there and computes peak intensities and profile parameters, and scales the intensities with the scale factor S.
The product Z ∙ M in the equation for quantification given above represents the mass of all atoms in the unit cell. Since we don’t know anything about the content of the unit cell in the unknown structure, we must provide the density of the phase in g/cm3 instead. From this value, the algorithm can reconstruct the mass per unit cell volume V, i.e. the product of Z ∙ M, and substitute it in the equation above. In summary, if the atomic site information of a specific phase is unknown, we can instead create a structure file containing the following information:
- Space group
- Cell parameters
- Calibrated structure factors Fhkl of the pure phase
- Density
The space group and cell parameters are used to calculate the peak positions, pre-calculated structure factors are used to calculate peak intensities, and the density is needed to calculate the phase quantity. The space group, cell parameters and density can usually be obtained by identifying the phase with a hkl line database such as the ICDD PDF-2. The list of structure factors Fhkl, however, must be determined experimentally.
This tutorial describes how to obtain an Fhkl list for a nano-crystalline apatite phase (Ca5(PO4)3OH), and how to use it to quantify apatite in a mixture with β-TCP (β-Ca3(PO4)2) without using the atomic site information of the apatite structure.
Preparations
The following materials are required:
- Pure unknown phase (here: Hydroxyapatite)
- An internal standard (here: NIST SRM 676a corundum)
- One or several mixtures of β-TCP and apatite in an unknown ratio
The pure apatite phase must be mixed in a 50:50 ratio by weight with the internal standard. We call this the reference sample. Then measure the reference sample and all β-TCP-apatite mixtures with the same instrument setup.
Creating the Fhkl structure file
Load the reference dataset (apatite + corundum) in Profex and create a refinement project with the phases Corundum and Apatite-OH. (The example files were measured with the instrument configuration RMS-D8-ADS-15-LynxEyeXE.) Then open the Apatite-OH.str file and modify it as follows, because we want to pretend that the structure is unknown and we need to use a LeBail fit:
PHASE=Hydroxyapatite // 01-074-0565 MineralName=Hydroxylapatite // Formula=Ca5_(PO4)3_(OH) // SpacegroupNo=176 HermannMauguin=P6_3/m // PARAM=A=0.9424_0.9330^0.9518 PARAM=C=0.6879_0.6810^0.6948 // RP=4 k1=0 PARAM=k2=0_0^0.0001 B1=ANISO^0.05 GEWICHT=0 // density=3.1654 GOAL=GrainSize(0,0,1) // GOAL=GrainSize(1,0,0) // GOAL:Hydroxyapatite=GEWICHT*ifthenelse(ifdef(d),exp(my*d*3/4),1) LeBail=1
Delete all lines starting with E= at the bottom of the file. Then run the refinement. We should obtain a good fit of the corundum and apatite peaks (Figure 1).
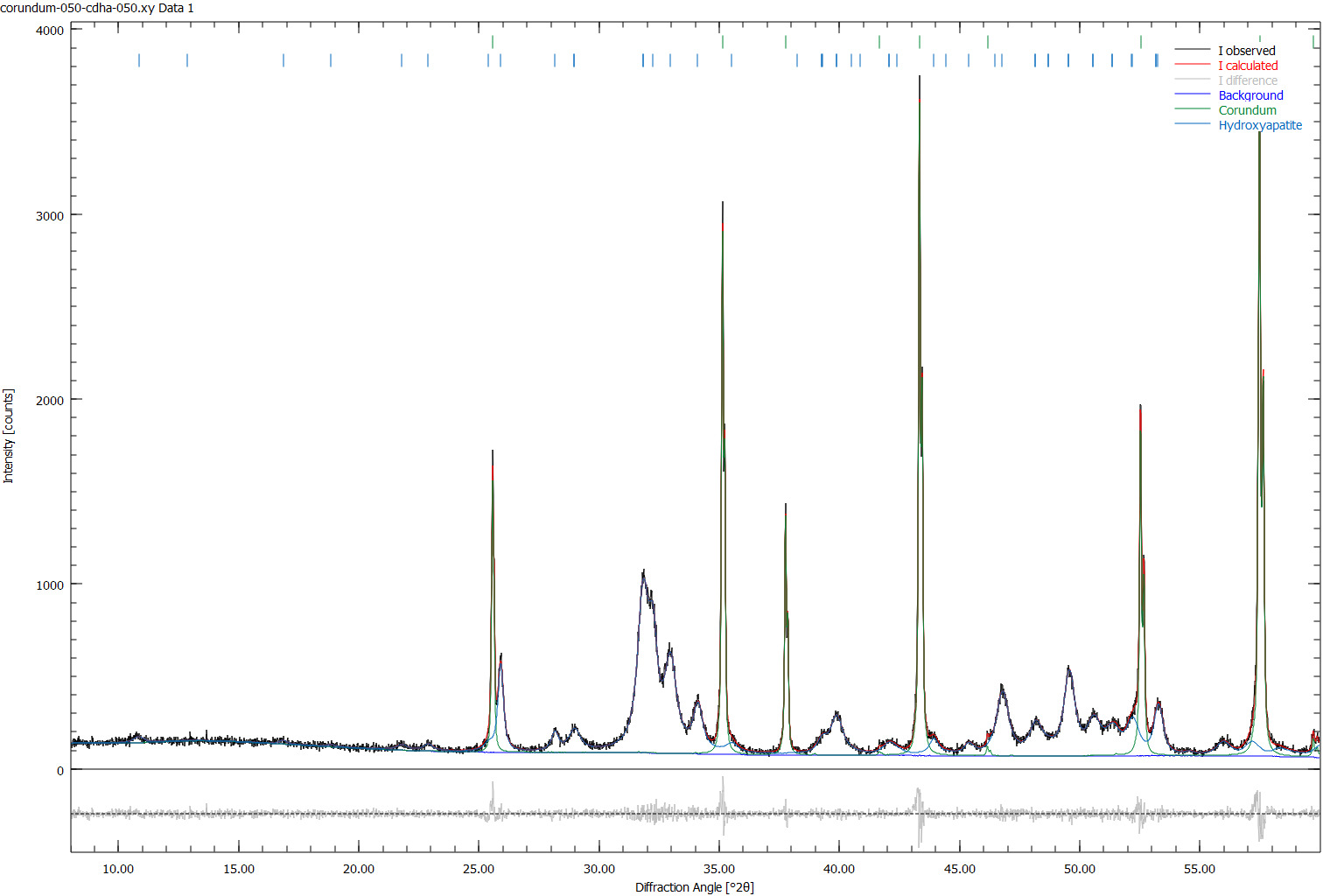
The GEWICHT parameter cannot be determined in LeBail mode. However, since we created a 50:50 mixture with corundum, we know that apatite must have the same GEWICHT as corundum. Once the refinement has completed, go to the *.lst file and locate the GEWICHT value of corundum:
GEWICHT=SPHAR4, MeanValue(GEWICHT)=0.0725296
Transfer this value to the apatite structure file. Do not refine GEWICHT of apatite, it must be hard-coded:
RP=4 k1=0 PARAM=k2=0_0^0.0001 B1=ANISO^0.05 GEWICHT=0.0725296 //
Then we instruct BGMN to write a new structure file with Fhkl values instead of atomic coordinates. Go to the refinement control file *.sav, right-click on the STRUC[2]=Apatite-OH.str entry, and select “Add SimpleSTRUCOUT file” from the menu (Figure 2). Then repeat the refinement. The resulting fit should look exactly as before (Figure 1), but a SimpleSTRUCOUT file was created.

Right-click on the “SimpleSTRUCOUT[2]=…” line and select “open file as text” to open the new Fhkl structure file. It looks like this (truncated):
PHASE=Hydroxyapatite MineralName=Hydroxylapatite Formula=Ca5_(PO4)3_(OH) SpacegroupNo=176 HermannMauguin=P6_3/m PARAM=A=9.40735424114519E-01_0.9330^0.9518 PARAM=C=6.87501861804558E-01_0.6810^0.6948 RP=4 k1=0 PARAM=k2=7.08917315231636E-06_0^0.0001 B1=ANISO^0.05 GEWICHT=0.0725296 density=3.1654 GOAL=GrainSize(0,0,1)=42.0 GOAL=GrainSize(1,0,0)=15.45 GOAL:Hydroxyapatite=GEWICHT*ifthenelse(ifdef(d),exp(my*d*3/4),1) LeBail=1 FMult=1 density=3.1654 my=0.000000 PARAM=GEWICHT=0_0 F=ifthenelse(ifdef(F[iref,h,k,l]),F[iref,h,k,l],0) F[1,1,-1,0]=19.0608 F[1,0,0,1]=7.2469 … F[1,6,-4,0]=62.0273 // You may want to remove parts or all of the below lines B1[1]=ifthenelse(ifdef(B10[h,k,l]),B10[h,k,l],0) B10[1,-1,0]=0.027467 … B10[6,-4,0]=0.027467 k1[1]=0 k2[1]=0.0000070892
Because we provided GEWICHT and density, BGMN was able to calibrate the amplitude of all structure factors F[h,k,l]. Before we can use this structure file for quantification of unknown apatite contents, we must modify it manually as follows (the sections are highlighted above):
- Scroll down half way and locate the following comment line:
// you may want to remove parts or all of the below lines
Delete everything from this line to the end of the file. - Delete the line
GEWICHT=0.0725296 - Delete the line
LeBail=1 - Delete
my=0.000000
Then save the file under a new name, for example “Apatite-OH-Fhkl.str”. It can now be used to quantify apatite in the unknown mixtures with β-TCP.
Using the Fhkl file
We want to use the Apatite-OH-Fhkl.str file to quantify apatite in the three mixtures with β-TCP provided in the example files. We have to add the apatite structure file manually instead of using the “Add/Remove phase” dialog (+- icon), because it is not yet in the structure file repository:
- Copy the file “Apatite-OH-Fhkl.str” from the “reference” folder to the “unknown” folder, so we can use it in the new refinement projects.
- Open the datasets of the unknown mixtures of b-TCP and Apatite-OH in Profex.
- Create a new refinement project by adding the phase “TCP-beta.str” and the instrument configuration “RMS-D8-ADS-15-LynxEyeXE”.
- Then modify the refinement control file *.sav to add the new apatite Fhkl structure file to the refinement. Add the highlighted parts manually:
% SampleID: cdha-025-bTCP-075.xy Data 1 % Theoretical instrumental function VERZERR=RMS-D8-ADS-15-LynxEyeXE.geq % Wavelength LAMBDA=CU % Phases STRUC[1]=TCP-beta.str STRUC[2]=Apatite-OH-Fhkl.str % Measured background UNT=RMS-D8-ADS-15-LynxEyeXE-bkgr.xy … sum=betaTCP+Hydroxyapatite QbetaTCP=betaTCP/sum QHydroxyapatite=Hydroxyapatite/sum GOAL[1]=QbetaTCP GOAL[2]=QHydroxyapatite
Then run the refinement. You can also use the button “Copy the current control file to all open projects” and run a batch refinement to refine all three example files in a row.
After the refinement, the quantities of both phases will be shown in the “Refined Parameters” window (Figure 3), and the results should match with the quantities given in the file names (“cdha” stands for “apatite”).
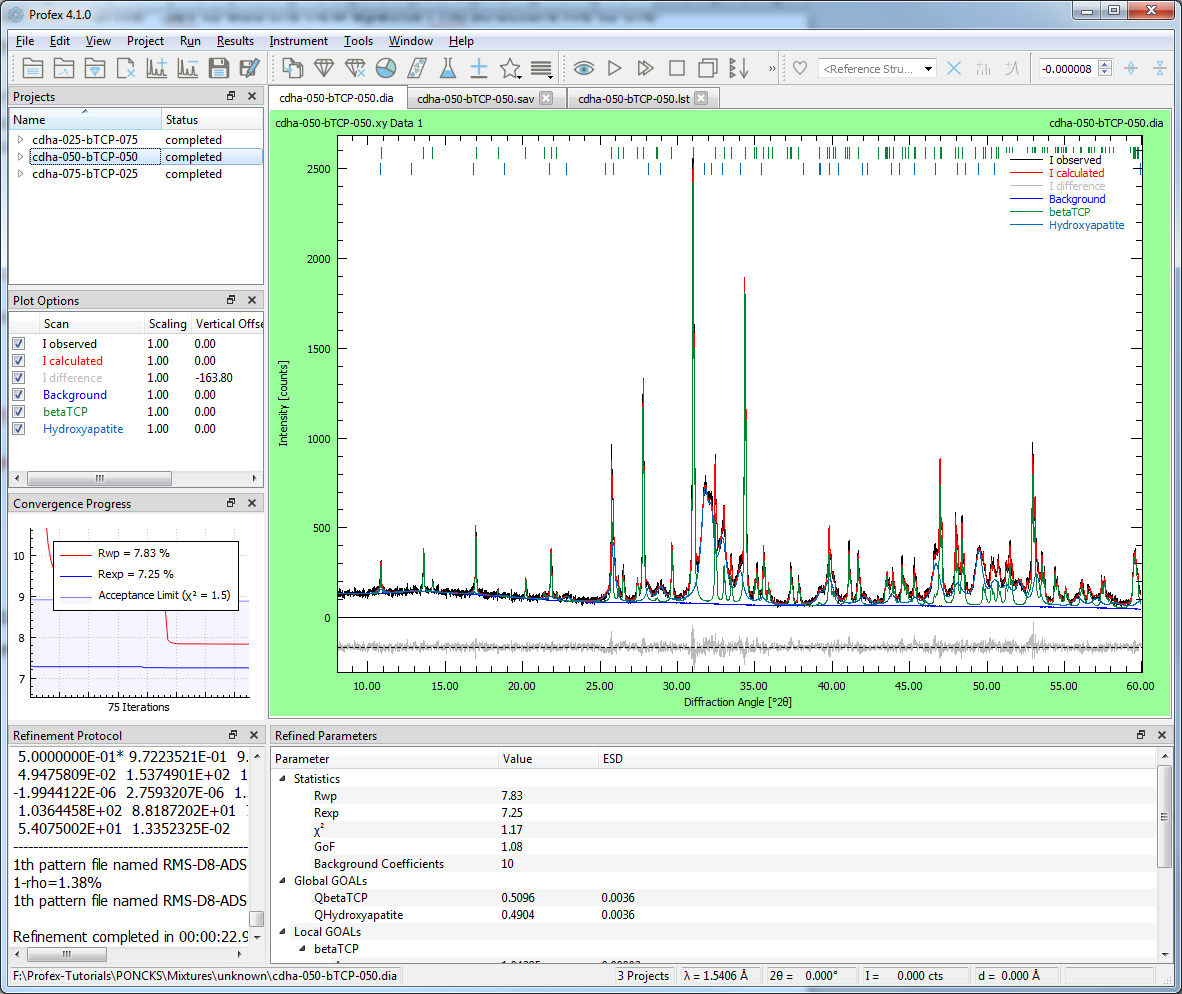
Improving the fit
The quantification can further be improved by changing the β-TCP structure file as follows:
- Refine bimodal crystallite sizes (β-TCP was somewhat over-milled).
- Refine Mg substitution on the sites Ca4 and Ca5.
In the solutions to the example files provided with this tutorial, the β-TCP and corundum structure refinements are already optimized.
References
Nicola Döbelin, April 2020